Cystic Fibrosis in Children
What is Cystic Fibrosis?
Cystic fibrosis is a genetic or inherited condition which is now increasingly recognized in the Indian sub-continent.
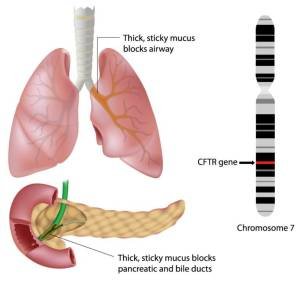
Cystic fibrosis is caused by a defect in the cystic fibrosis transmembrane conductance regulator (CFTR) gene. Cystic fibrosis is inherited in an autosomal recessive manner. Presence of mutations in both copies of the gene for the cystic fibrosis transmembrane conductance regulator (CFTR) protein leads to cystic fibrosis disease. Parents who have a single working copy are carriers and otherwise mostly healthy.
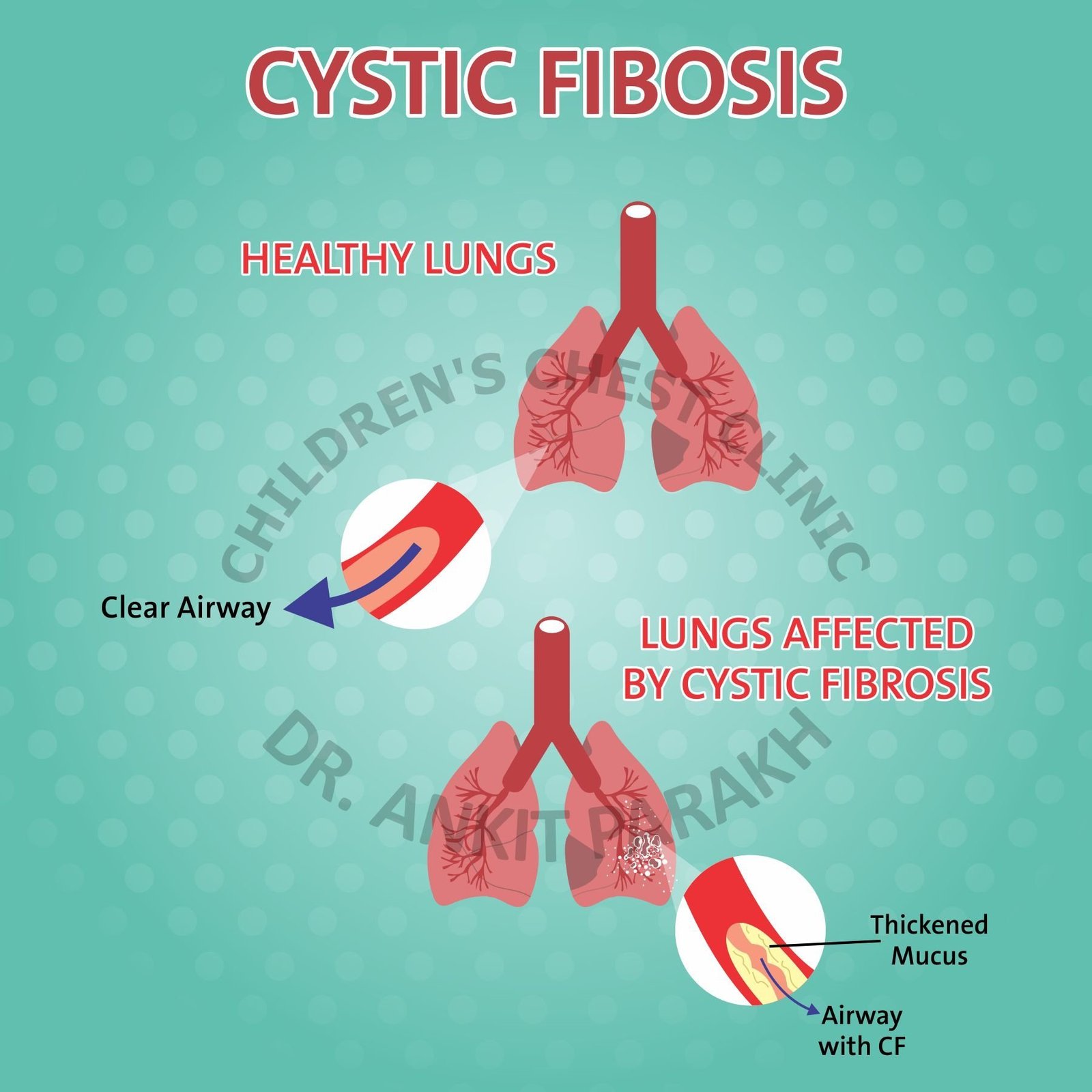
Cystic fibrosis transmembrane conductance regulator (CFTR) is involved in the production of sweat, digestive fluids, and mucus. When the CFTR defective, secretions become thick and sticky. This leads to damage to multiple organs of the body but mainly the lungs and digestive system.
Symptoms of Cystic Fibrosis in Children
Symptoms of Cystic Fibrosis can vary but commonly include:
- Persistent wet coughing, sometimes with phlegm/sputum
- Frequent lung infections, such as pneumonia
- Wheezing or shortness of breath
- Poor growth or weight gain despite a healthy appetite
- Salty-tasting skin (due to excessive salt in their sweat)
- Difficulty with bowel movements or frequent greasy stools
Diagnosis
For diagnosis of cystic fibrosis following tests might be required:
- Sweat Chloride Test: Sweat Chloride analysis is the standard investigation to confirm cystic fibrosis across the world. It involves collection of sweat of the child with a Gibson & Cooke’s method followed by chloride estimation in the sweat. Value of chloride in the sweat more than 60 mmol/L are suggestive of cystic fibrosis. For more on sweat chloride analysis click here.
- Genetic Tests: Genetic Testing is available which looks for mutations in the CFTR gene which is responsible for regulating chloride transport in the lungs, pancreas, and gut.
Treatment Options for Cystic Fibrosis
Advancements in CF care have significantly improved outcomes. Treatment typically includes:
- Mucus-thinning drugs to clear airways (hypertonic saline, DNAse)
- Antibiotics to prevent and treat infections
- CFTR modulators that target the underlying genetic defect
- Chest physiotherapy to loosen mucus
- Devices like Acapella, Aerobika
- Enzyme supplements (Creon) to aid digestion
- High-calorie diets and vitamin supplements
- Lung function tests
- Growth and nutrition assessments
- Lung transplants in severe cases
Cystic Fibrosis is a challenging condition requiring lifelong management, but with early diagnosis, comprehensive care, and recent medical advancements, children with Cystic Fibrosis can live healthier and more active lives. If you suspect your child may have symptoms of Cystic Fibrosis or if your family has a history of the condition, consult a pediatric pulmonologist to discuss testing and treatment options.
Conclusion
Related Video
Frequently Asked Questions (FAQs)
1. What are the early signs of Cystic Fibrosis in children?
Early signs include salty-tasting skin, persistent coughing, and difficulty gaining weight despite a healthy appetite.
2. What happens to the lungs in Cystic Fibrosis?
Lungs are the primary organs involved in cystic fibrosis. In children with cystic fibrosis the mucus gets thick, viscid and sticky. These secretions plug-up the small airways of the lungs. The small plugged airways get repeated infections and lead to chronic lung damage over time. These problems begin very early in life starting from the first few months of life.
3. What other organs are affected in children with Cystic Fibrosis?
Cystic fibrosis also affects the organs of the digestive system like the liver, pancreas and intestines. This leads to reduced and ineffective digestive juice leading to impaired food absorption. Hence these children would have large, bulky and frothy stools. Because of poor food absorption children get poor weight gain.
4. How is Cystic Fibrosis treated in children?
Treatment involves a combination of medications, airway clearance techniques, nutritional support, and regular monitoring to manage symptoms and prevent complications.
5. Can Cystic Fibrosis be cured?
Currently, there is no cure for Cystic fibrosis. However, advances in therapy, including CFTR modulators, are improving quality of life and longevity.